Welcome to CIDPUSA.ORG
Autoimmune Facts - for autoimmune diseases
November 19, 2021
alternatives treatment of autoimmune disease read our e-book
DeLisa Fairweather and Noel R. Rose*
*Johns Hopkins University, Baltimore, Maryland, USA.
Autoimmune diseases are the number one category of disease in the United States cancer and heart disease are triggered by autoimmune process; they affect approximately 5%-8% of the population or 14-22 million persons . Autoimmune diseases can affect virtually every site in the body, including the endocrine system, connective tissue, gastrointestinal tract, heart, skin, and kidneys. At least 15 diseases are known to be the direct result of an autoimmune response, while circumstantial evidence implicates >80 conditions with autoimmunity . In several instances, such as rheumatoid arthritis, multiple sclerosis, and myocarditis, the autoimmune disease can be induced experimentally by administering self-antigen in the presence of adjuvant (collagen, myelin basic protein, and cardiac myosin, respectively) . An important unifying theme in autoimmune diseases is a high prevalence in women . Conservative estimates indicate that 6.7 million or 78.8% of the persons with autoimmune diseases are women .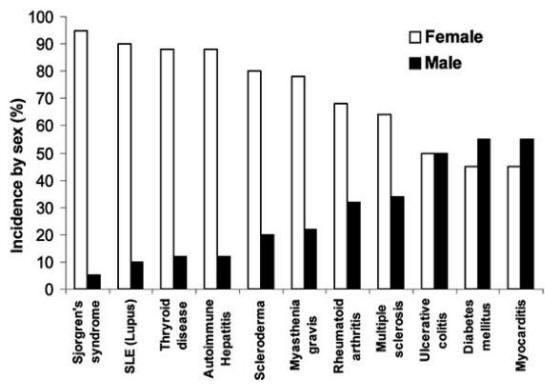
Soon after autoimmune diseases were first recognized more than a century ago, researchers began to associate them with viral and bacterial infections. Autoimmune diseases tend to cluster in families and in individuals (a person with one autoimmune disease is more likely to get another), which indicates that common mechanisms are involved in disease susceptibility. Studies of the prevalence of autoimmune disease in monozygotic twins show that genetic as well as environmental factors (such as infection) are necessary for the disease to develop. Genetic factors are important in the development of autoimmune disease, since such diseases develop in certain strains of mice (e.g., systemic lupus erythematosus or lupus in MRL mice) without any apparent infectious environmental trigger. However, a body of circumstantial evidence links diabetes, multiple sclerosis, myocarditis, and many other autoimmune diseases with preceding infections . More often, many different microorganisms have been associated with a single autoimmune disease, which indicates that more than one infectious agent can induce the same disease through similar mechanisms . Since infections generally occur well before the onset of symptoms of autoimmune disease, clinically linking a specific causative agent to a particular autoimmune disease is difficult . This difficulty raises the question of whether autoimmune diseases really can be attributed to infections.

Read presidentsKennedy's autoimmune history
for a Help yourself please read the diet section