Multifocal motor neuropathy subtype
For treatments of
autoimmune disorders please read our
e-book
Sat, 31 May 2008 17:55:07
By Patricia Khashayar, MD.,
Types of Lewis Sumner
Lewis-Sumner syndrome
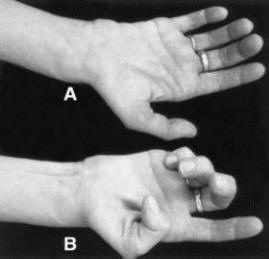
A)Trying to straighten the fingers
(B)trying to make a fist but weakness is obvious in LSS.
Many docs misdiagnose as Carpal tunnel or Ulnar neuropathy
RETURN TO MAIN PAGE OF L.S.S
Rinsho Shinkeigaku.
1999 Jan;39(1):107-9.
Diagnosis and treatment of multifocal motor neuropathy (Lewis-Sumner)
Kaji R.
Department of Neurology, Kyoto University Hospital.
We made a retrospective long-term follow-up study of 25 patients with multifocal motor neuropathy (Lewis-Sumner). The diagnosis was based upon criteria modified from those of AAEM (Sumner 1997). The electrophysiological findings indicating conduction block or focal demyelinative lesions were more diagnostic than anti-GM 1 antibody titers, which were elevated in only 40% of these patients. Demonstration of definite conduction block was not always possible in those patients who responded favorably to intravenous immunoglobulins (IVIg), whereas indirect pieces of evidence such as F-wave abnormalities or focal conduction delay or dispersion were equally helpful. IVIg had superior outcome to cyclophosphamide, which sometimes caused serious adverse effects. Three patients with severe axonal involvement showed elevated monospecific antibodies to GalNAc-GD1a.
-
| Brain. 2004 Sep;127(Pt 9):2010-7. Epub 2004 Aug 2.
Follow-up study and response to treatment in 23 patients with Lewis-Sumner syndrome.
Lewis-Sumner syndrome (LSS) is a dysimmune peripheral nerve disorder, characterized by a predominantly distal, asymmetric weakness mostly affecting the upper limbs with sensory impairment, and by the presence of multifocal persistent conduction blocks. The nosological position of this neuropathy in relation to multifocal motor neuropathy (MMN) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is still debated. We report the clinical, biological and electrophysiological features, the course and the response to treatment in 23 LSS patients. The initial symptoms started in the distal part of an upper limb in 70% of patients. They were sensorimotor in 65% and purely sensory in 35% of patients. A cranial nerve involvement was observed in 26% of patients and a distal limb amyotrophy in 52%. The CSF protein level was normal in 67% of patients and mildly elevated in the remainder. None had serum anti-GM1 antibodies.
There were multiple motor conduction blocks (average of 2.87/patient), predominantly located in the forearm, whereas demyelinating features outside the blocked nerves were rare. Abnormal distal sensory potentials were found in 87% of patients. The electrophysiological pattern suggests a very focal motor fibre demyelination sparing the nerve endings, whereas sensory fibre involvement was widespread. The course was chronic progressive in 71% of patients and relapsing-remitting in the others. During the follow-up study (median duration of 4 years), half of the patients progressed with a multifocal pattern and the distribution of the motor deficit remained similar to the initial presentation. The other patients showed a progression to the other limbs, suggesting a more diffuse process. Fifty-four percent of the patients treated with intravenous immunoglobulin showed an improvement, compared with 33% of the patients treated with oral steroids. Overall, 73% of patients had a positive response to immune-mediated therapy. LSS may be distinguished from MMN by the presence of sensory involvement, the absence of serum anti-GM1 antibodies and, in some cases, a positive response to steroids. In some of the patients in our study, LSS evolved into a more diffuse neuropathy sharing similarities with CIDP. Others had a clinical course characterized by a striking multifocal neuropathy, which suggests underlying mechanisms different from CIDP. Overall, whatever the clinical course, LSS responded to immune-mediated treatment in a manner similar to CIDP.
PMID: 15289267 [PubMed - indexed for MEDLINE]
| |
Return to Lewis Sumner Page